10x scRNASeq
This nf-core pipeline does RNA sequencing analysis using cellranger count.
Please follow the instructions below to get access to our analysis pipeline and run it on your scRNAseq data:
Prerequisites:
CCBB assumes that you have
- Signed up to request access to the pipeline. If not, please sign up using this form
- an account on Garibaldi,
- set up Workflow on your computer. If this is not setup, please follow the instructions to set it up.
Step1:
On Garibaldi, create a folder with your sample fastqs
- Sample fastqs need to be gzipped with extension .fastq.gz
- In the same folder, generate “samplesheet.csv” files based on the examples provided here.
- Edit them to reflect your sample name, /full-path-to-bulk-RNAseq-folder-on-Garibaldi data files (fastqs)
Sample Naming Conventions
Please follow the sample naming conventions listed to avoid errors. The fastq name should be SampleName_SampleNumber_LaneNumber_R1_001.fastq.gz and SampleName_SampleNumber_LaneNumber_R2_001.fastq.gz.
Examples - sample1_S1_L001_R1_001.fastq.gz and sample1_S1_L001_R2_001.fastq.gz, sample2_S2_L001_R1_001.fastq.gz and sample2_S2_L001_R2_001.fastq.gz
- SampleName should be a single word and unique for each sample with no special characters (space, #, _ etc.)
- SampleNumber should be “S” followed by a number - unique for each sample with no special characters (space, #, _ etc.)
- If the samples are sequenced across multiple lanes, make sure to provide the same SampleName
Step2:
Now you are ready to run the CCBB scRNASeq analysis!
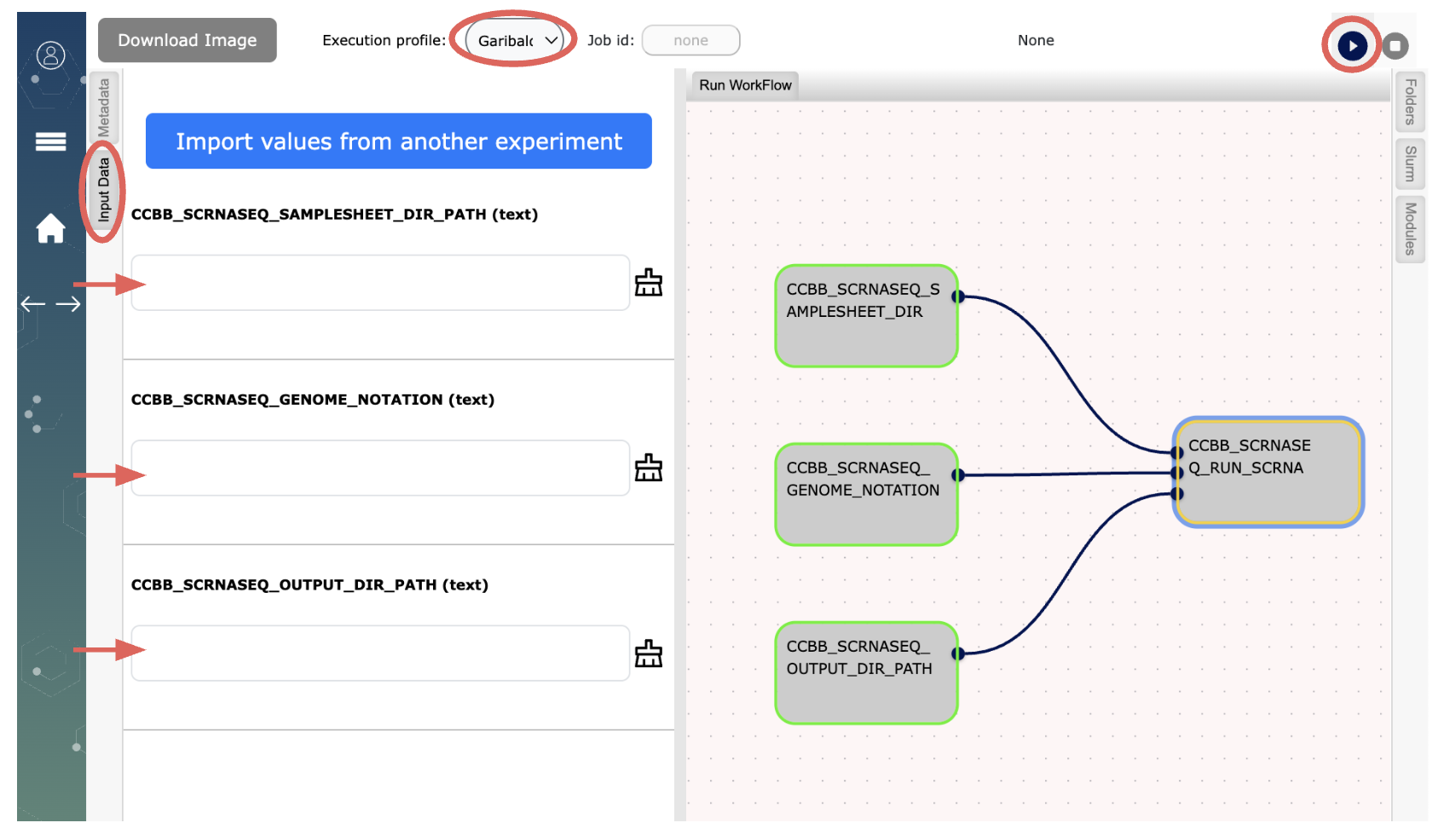
Follow the instructions below to begin: Open Workflow on your browser (Refer to the image at the end of this document for details)
- Click on “Experiments”
- To select a workflow click on “My WorkFlows” (on the top right corner), then click on “New Experiment”
- Click on “CCBB_SCRNASeq_WF”
- Choose Garibaldi from the dropdown menu for “Execution profile:” (top of the page)
The workflow “CCBB_SCRNASeq_WF” is used to run the scRNASeq pipeline. This needs three inputs as defined below.
- Click on the “Input Data” tab and
- (a) type in the full path to your folder on Garibaldi with samplesheet.csv,
- (b) type in the reference genome code for your samples (e.g. HU0 (zero not the letter O) for human, MM0 for mouse
- (c) type in the full path to where you want the results of the scRNASeq analysis to be generated on Garibaldi.
IMPORTANT NOTE ON GENOMES:
Currently the pipeline will work only for Human, Mouse.
If you have any other custom species please contact CCBB
Follow the details to understand the pipeline outcome for your data.
Step3:
Post analysis follow-up for users.
Please follow the instructions that are sent via email upon pipeline completion:
- Important cleanup step: After the successful completion of the scRNASeq pipeline, please remove the “work” sub-folder (that holds the intermediate files/folders generated by the pipeline) in your workflow setup folder on Garibaldi (fullpath given in the email).
- Contact CCBB for any questions regarding the pipeline, results of analysis, further assistance with downstream analysis (like pathway analysis, custom differential expression analysis, specific plots for publication etc.) and your feedback on our pipeline !
- Please take a few minutes to give us your feedback
NOTE:
- The job completion time depends on various factors such as number of samples, depth of sequencing etc.
- The job runs on a node on garibaldi. Depending on the cluster capacity, there may be a wait time to obtain the node.
DISCLAIMER
CCBB is providing pipelines on Garibaldi, the institute’s shared linux cluster, on “as is” and “as available” terms, solely for the benefit of the scientific community at Scripps Research.
CCBB reserves the right to continue/discontinue any of its pipelines on Garibaldi.