RNASeq Pipeline
RNA-seq data is valuable as it allows the measure of RNA expression levels as a transcriptional readout and the study of RNA structures in order to understand how RNA-based mechanisms impact gene regulation and thus disease and phenotypic variation encodeproject
rRNA databases:
Reference build:
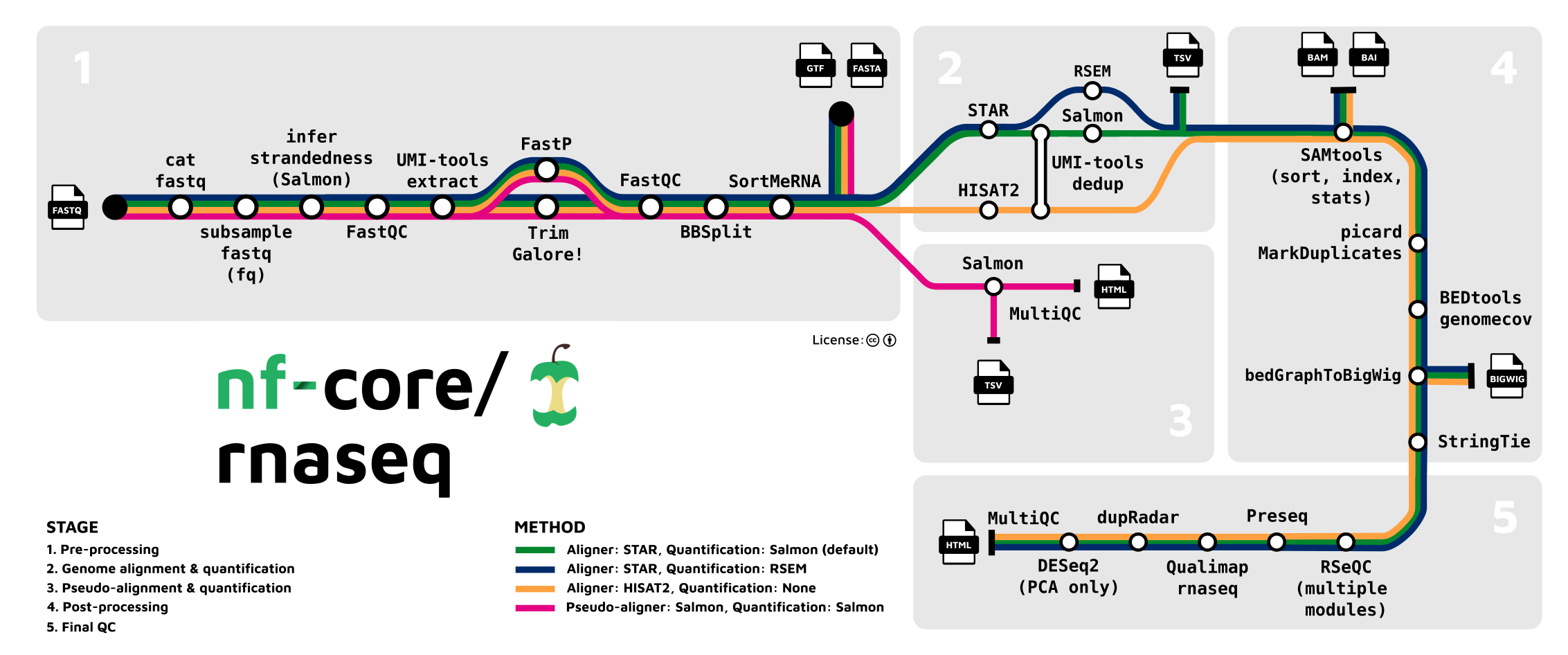
Steps in data processing:
| Step | Software/Module | Input | Output |
|---|---|---|---|
| Assess Data Quality | fastqc | *.fastq.gz files | websummary.html |
| Adapter and Quality Trimming of Reads | TrimGalaore! | *.fastq.gz files | Trimmed *.fastq.gz files |
| Removal of Ribosomal RNA | SortMeRNA | Trimmed *.fastq.gz files; rRNA databases | Ribosomal RNA removed and trimmed *.fastq.gz files |
| Alignment to the Genome | STAR | Ribosomal RNA removed and trimmed *.fastq.gz files; reference build | *.bam files |
| Sort and index alignment | SAMTools | *.bam files | Sorted .bam files and .bai files |
| Duplicate Read Marking | Picard markDuplicates | Sorted .bam files and .bai files | .markDups.bam files and .markDups.bai files |
| Quality control | MultiQC | Output summaries from RSeQC, Qualimap, dupRadar, Preseq, edgeR | websummary.html |
| Expression quantification | featureCounts | .markDups.bam files and .markDups.bai files; reference build | *.featureCounts.txt files |
| Differential Expression | DESeq2 | *.featureCounts.txt files; sample metadata (names, groups, contrasts) | deseq2.results.txt files and *.deseq2.plots.pdf files |